
Infer presence and mode of polyploidy
Below are the inputs, commands, and outputs to do several analyses with GRAMPA. The inputs are based on simulated data. For more detailed info on the simulations check our paper.
The examples below call GRAMPA as grampa assuming it has been installed from bioconda. If you installed
from source, see usage details in the README.
Allopolyploidy
Inputs
1000 gene trees were simulated with gain and loss using JPrIME based on the following allopolyploid-like MUL-tree:
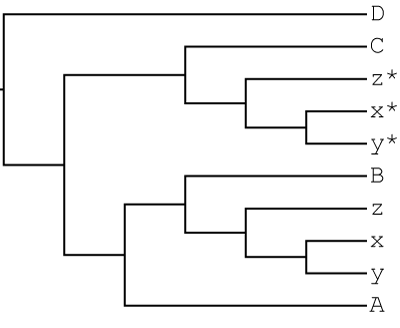
In this scenario, lineages B and C hybridized to form an allopolyploid lineage that diversified into the x,y,z clade.
We then remove one polyploid clade from the MUL-tree to get a singly-labeled tree as input for GRAMPA. This is the same topology as above, except that the x,y,z clade sister to C is removed. This is the type of tree that typical phylogenetic reconstruction programs would produce even in the presence of allopolyploidy:
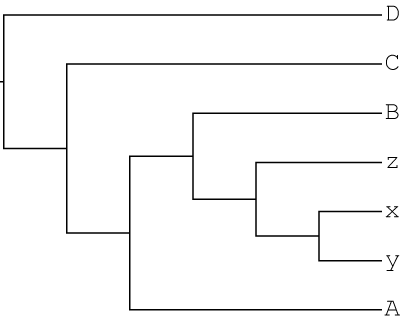
So, the input files for this search are:
- Singly-labeled species tree: spec_tree_3a.tre
- 1000 gene trees simulated from an allopolyploid MUL-tree: gene_trees_3a.txt
GRAMPA command
Since in reality we wouldn't know whether there is an allo-, auto-, or no polyploidy in this tree, we want GRAMPA to search all nodes as possible
polyploid lineages. That means we don't specify -h1 or -h2.
grampa -s spec_tree_3a.tre -g gene_trees_3a.txt -o allo-example-output -f allo-testOutputs
The above command would create the directory allo-example-output with six output files
- allo-test-checknums.txt
- allo-test-detailed.txt
- allo-test-dup-counts.txt
- allo-test.log
- allo-test-scores.txt
- allo-test-trees-filtered.txt
Since we are trying to determine the mode of polyploidy, we are interested in the allo-test-scores.txt file. This file contains
the total reconciliation scores for each MUL-tree considered, sorted from lowest scoring tree to highest scoring tree, and looks something like this:
mul.tree h1.node h2.node score labeled.tree
105 <2> C 5018 ((((((x+,y+)<1>,z+)<2>,B)<3>,A)<4>,(C,((x*,y*)<5>,z*)<6>)<7>)<8>,D)<9>
113 <3> C 5900 ((((((x+,y+)<1>,z+)<2>,B+)<3>,A)<4>,(C,(((x*,y*)<5>,z*)<6>,B*)<7>)<8>)<9>,D)<10>
119 <4> C 6824 ((((((x+,y+)<1>,z+)<2>,B+)<3>,A+)<4>,(C,((((x*,y*)<5>,z*)<6>,B*)<7>,A*)<8>)<9>)<10>,D)<11>
122 <4> <5> 6824 (((((((x+,y+)<1>,z+)<2>,B+)<3>,A+)<4>,C)<5>,((((x*,y*)<6>,z*)<7>,B*)<8>,A*)<9>)<10>,D)<11>
110 <2> <5> 7306 (((((((x+,y+)<1>,z+)<2>,B)<3>,A)<4>,C)<5>,((x*,y*)<6>,z*)<7>)<8>,D)<9>
109 <2> <4> 7435 (((((((x+,y+)<1>,z+)<2>,B)<3>,A)<4>,((x*,y*)<5>,z*)<6>)<7>,C)<8>,D)<9>
117 <3> <5> 7616 (((((((x+,y+)<1>,z+)<2>,B+)<3>,A)<4>,C)<5>,(((x*,y*)<6>,z*)<7>,B*)<8>)<9>,D)<10>
GRAMPA tells us MUL-tree 105 is the lowest scoring tree:
((((((x+,y+)<1>,z+)<2>,B)<3>,A)<4>,(C,((x*,y*)<5>,z*)<6>)<7>)<8>,D)<9>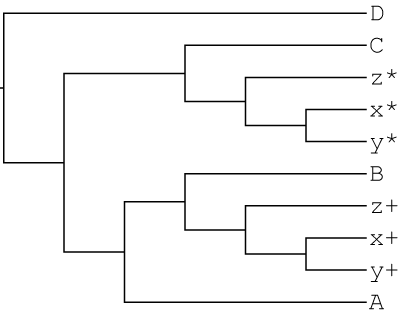
Notice that this is the same topology that was used to simulate the gene-trees. GRAMPA has successfully identified an allopolyploid MUL-tree and placed the second polyploid lineage on the correct branch!
Autopolyploidy
1000 gene trees were simulated with gain and loss using JPrIME based on the following autopolyploid-like MUL-tree:
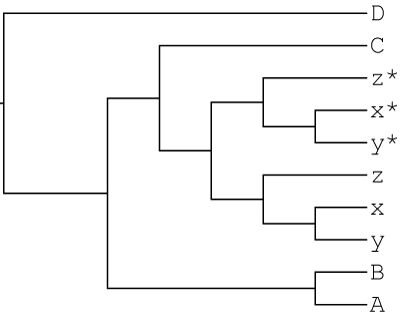
In this scenario, a lineage sister to species C underwent autopolyploidization and subsequently diversified into the x,y,z clade.
We then remove one polyploid clade from the MUL-tree to get a singly-labeled tree as input for GRAMPA. This is the same topology as above, except that one x,y,z clade is removed. This is the type of tree that typical phylogenetic reconstruction programs would produce even in the presence of autopolyploidy:
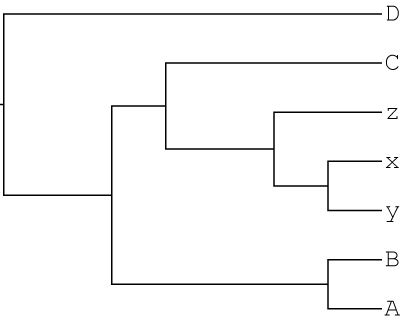
So, the input files for this search are:
- Singly-labeled species tree: spec_tree_18.tre
- 1000 gene trees simulated from an allopolyploid MUL-tree: gene_trees_18.txt
GRAMPA command
Since in reality we wouldn't know whether there is an allo-, auto-, or no polyploidy in this tree, we want GRAMPA to search all nodes as possible
polyploid lineages. That means we don't specify -h1 or -h2.
grampa -s spec_tree_18.tre -g gene_trees_18.txt -o auto_example_output -f auto_test -v 0Outputs
The above command would create the directory auto_example_output with six output files
- auto-test-checknums.txt
- auto-test-detailed.txt
- auto-test-dup-counts.txt
- auto-test.log
- auto-test-scores.txt
- auto-test-trees-filtered.txt
Since we are trying to determine the mode of polyploidy, we are interested in the auto-test-scores.txt file. This file contains
the total reconciliation scores for each MUL-tree considered, sorted from lowest scoring tree to highest scoring tree, and looks something like this:
mul.tree h1.node h2.node score labeled.tree
119 <3> <3> 4807 (((B,A)<1>,((((x+,y+)<2>,z+)<3>,((x*,y*)<4>,z*)<5>)<6>,C)<7>)<8>,D)<9>
0 NA NA 5476 (((B,A)<1>,(((x,y)<2>,z)<3>,C)<4>)<5>,D)<6>
85 D D 5493 (((B,A)<1>,(((x,y)<2>,z)<3>,C)<4>)<5>,(D+,D*)<6>)<7>
105 <2> z 5776 (((B,A)<1>,(((x+,y+)<2>,(z,(x*,y*)<3>)<4>)<5>,C)<6>)<7>,D)<8>
110 <2> <3> 5776 (((B,A)<1>,((((x+,y+)<2>,z)<3>,(x*,y*)<4>)<5>,C)<6>)<7>,D)<8>
71 C C 5812 (((B,A)<1>,(((x,y)<2>,z)<3>,(C+,C*)<4>)<5>)<6>,D)<7>
97 <1> <1> 5817 ((((B+,A+)<1>,(B*,A*)<2>)<3>,(((x,y)<4>,z)<5>,C)<6>)<7>,D)<8>
GRAMPA tells us MUL-tree 119 is the lowest scoring tree:
(((B,A)<1>,((((x+,y+)<2>,z+)<3>,((x*,y*)<4>,z*)<5>)<6>,C)<7>)<8>,D)<9>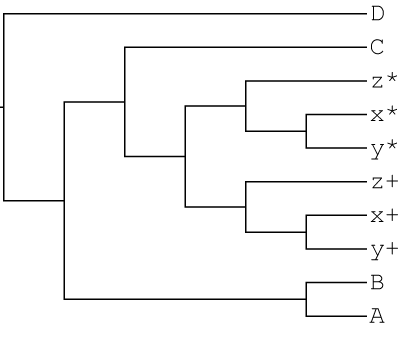
Notice that this is the same topology that was used to simulate the gene-trees, and that the h1 and h2 nodes of this MUL tree are the same, indicating autopolyploidy. GRAMPA has successfully identified an autopolyploid MUL-tree on the correct branch!
No polyploidy
1000 gene trees were simulated with gain and loss JPrIME based on the following singly-labeled tree:
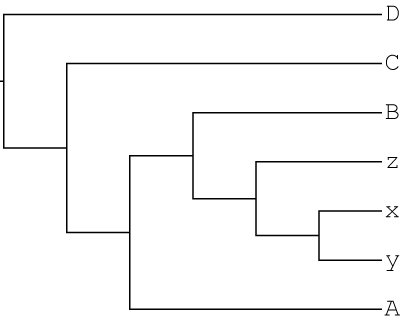
In this scenario, no polyploidy has occurred and this is the same tree we give to GRAMPA.
So, the input files for this search are:
- Singly-labeled species tree: spec_tree_33.tre
- 1000 gene trees simulated from an allopolyploid MUL-tree: gene_trees_33.txt
GRAMPA command
Since in reality we wouldn't know whether there is an allo-, auto-, or no polyploidy in this tree, we want GRAMPA to search all nodes as possible
polyploid lineages. That means we don't specify -h1 or -h2.
grampa -s spec_tree_33.tre -g gene_trees_33.txt -o nop_example_output -f nop_test -v 0Outputs
The above command would create the directory nop_example_output with four output files
- nop-test-checknums.txt
- nop-test-detailed.txt
- nop-test-dup-counts.txt
- nop-test.log
- nop-test-scores.txt
- nop-test-trees-filtered.txt
Since we are trying to determine the mode of polyploidy, we are interested in the nop-test-scores.txt file. This file contains
the total reconciliation scores for each MUL-tree considered, sorted from lowest scoring tree to highest scoring tree, and looks something like this:
mul.tree h1.node h2.node score labeled.tree
0 NA NA 4115 ((((((x,y)<1>,z)<2>,B)<3>,A)<4>,C)<5>,D)<6>
85 D D 4217 ((((((x,y)<1>,z)<2>,B)<3>,A)<4>,C)<5>,(D+,D*)<6>)<7>
71 C C 4370 ((((((x,y)<1>,z)<2>,B)<3>,A)<4>,(C+,C*)<5>)<6>,D)<7>
57 A A 4423 ((((((x,y)<1>,z)<2>,B)<3>,(A+,A*)<4>)<5>,C)<6>,D)<7>
43 B B 4554 ((((((x,y)<1>,z)<2>,(B+,B*)<3>)<4>,A)<5>,C)<6>,D)<7>
120 <4> D 4558 ((((((x+,y+)<1>,z+)<2>,B+)<3>,A+)<4>,C)<5>,(D,((((x*,y*)<6>,z*)<7>,B*)<8>,A*)<9>)<10>)<11>
113 <3> C 4632 ((((((x+,y+)<1>,z+)<2>,B+)<3>,A)<4>,(C,(((x*,y*)<5>,z*)<6>,B*)<7>)<8>)<9>,D)<10>
GRAMPA tells us that the input species tree (i.e. the singly-labeled tree) is the lowest scoring tree:
((((((x,y)<1>,z)<2>,B)<3>,A)<4>,C)<5>,D)<6>
Notice that this is the same topology that was used to simulate the gene-trees and contains no polyploidy events. GRAMPA has successfully determined that no polyploidy has occurred among these lineages!